

Introduction: DARA, a human IgGκ monoclonal antibody targeting CD38, is approved in combination with bortezomib, thalidomide and dexamethasone for transplant-eligible NDMM; however, RVd is the standard of care in the US for transplant-eligible NDMM. The phase 2 GRIFFIN study (NCT02874742) of DARA plus RVd (D-RVd) for transplant-eligible NDMM in the US included a safety run-in phase followed by a randomized phase. In the 16-patient safety run-in, no dose limiting toxicities (DLTs) occurred and no new safety concerns were identified for D-RVd (Voorhees P, Blood 2018 132 [Suppl 1]:151). The randomized phase of GRIFFIN met its prespecified primary endpoint with improved rate of stringent complete response (sCR) by the end of consolidation (D-RVd, 42.4% vs RVd, 32.0%; 1-sided P=0.068) (Voorhees P, Blood 2020); however, median follow-up was limited (13.5 months). Therefore, we conducted an updated analysis of efficacy and safety for the safety run-in cohort to examine longer follow-up data for D-RVd and evaluate the role of DARA plus lenalidomide (D-R) maintenance therapy.
Methods: Patients with NDMM eligible for high-dose therapy (HDT) and autologous stem cell transplant (ASCT) were enrolled. Patients in the safety run-in cohort received 4 induction cycles of D-RVd, HDT, ASCT, 2 consolidation cycles of D-RVd, and 24 months of maintenance with D-R. During induction and consolidation, patients received R 25 mg PO on Days 1‐14; V 1.3 mg/m2 SC on Days 1, 4, 8, and 11; and d 40 mg QW every 21 days. DARA 16 mg/kg IV was given on Days 1, 8, and 15 of Cycles 1‐4 and Day 1 of Cycles 5‐6. During maintenance (Cycles 7-32), patients received R 10 mg (15 mg in Cycles 10+ if tolerated) on Days 1‐21 every 28 days plus DARA 16 mg/kg IV Q8W (or Q4W per patient decision after protocol Amendment 2). The primary objective of the safety run-in phase was to assess DLTs. Additional endpoints included safety, sCR rate by the end of consolidation and end of maintenance per IMWG criteria by computer algorithm, progression-free survival (PFS), and minimal residual disease (MRD) negativity (10-5 and 10-6 threshold) assessed by next-generation sequencing (clonoSEQ; Adaptive Biotechnologies).
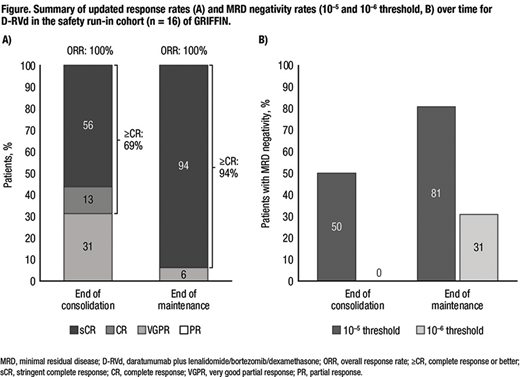
Results: Sixteen patients enrolled in the safety run-in and received D-RVd. Median age was 62.5 years, 8 (50%) patients were male, 4 (25%) were International Staging System stage II or III, and 4 (25%) had high cytogenetic risk based on fluorescence in situ hybridization. Thirteen (81%) patients completed study therapy, and 3 (19%) patients discontinued due to progressive disease (n=2) or adverse event (n=1). With continued therapy, responses further deepened for D-RVd (Figure). By the end of D-RVd consolidation, all patients responded, with a best response of sCR for 9 (56%) patients. By the end of D-R maintenance therapy, 15 (94%) patients achieved a best response of sCR. MRD negativity (10‒5) at the end of consolidation occurred in 8 patients (50%); no patient was MRD negative at 10-6. By the end of maintenance, 13 (81%) patients were MRD negative at 10‒5, and 5 (31%) were MRD negative at 10‒6. At a median follow-up of 39.0 months, 3 of 16 patients have progressed, with estimated 24-month and 36-month PFS rates of 94% and 78%, respectively. One death occurred, due to progressive disease in the patient who failed to achieve sCR. With longer follow-up, no new safety concerns were identified. Grade 3-4 treatment emergent adverse events (TEAEs) occurred in 14 (88%) patients, with the most common being neutropenia (44%; n=7), pneumonia (31%; n=5), lymphopenia (25%; n=4), and thrombocytopenia (25%; n=4). One patient had TEAEs leading to discontinuation of study treatment. No grade 5 TEAEs occurred. Infusion-related reactions occurred in 4 (25%) patients (all grade 1-2), mainly during the first cycle.
Conclusions: With >3 years of follow-up in the safety run-in cohort of GRIFFIN, D-RVd induction, ASCT, and consolidation led to durable responses that deepened with 2 years of D-R maintenance. There were no new safety concerns. These emerging data suggest that the deep responses seen in the randomized phase of GRIFFIN are durable and will continue to improve over time, and that D-RVd may be a potential new standard of care for transplant-eligible NDMM.
Support: Alliance Foundation Trials; https://acknowledgments.alliancefound.org; Janssen Oncology
Voorhees:Janssen: Other: Advisory Board; Novartis: Consultancy; Oncopeptides: Consultancy, Honoraria; TeneoBio: Other: Advisory Board; Adaptive Biotechnologies: Other: Advisory Board; BMS: Other: Advisory Board; GSK: Honoraria. Rodriguez:BMS, Takeda, Amgen: Consultancy, Speakers Bureau. Reeves:Bristol Myers Squibb: Speakers Bureau; Incyte: Honoraria; Takeda: Honoraria. Costa:Amgen: Consultancy, Honoraria, Research Funding; BMS: Consultancy, Honoraria; Genentech: Consultancy; Celgene: Consultancy, Honoraria; AbbVie: Consultancy; Sanofi: Consultancy, Honoraria; Janssen: Consultancy, Honoraria, Research Funding. Lutska:Janssen: Current Employment. Bobba:Janssen: Current Employment. Hoehn:Merck (current), Janssen (within 24 months): Current Employment, Ended employment in the past 24 months. Pei:Janssen: Current Employment, Current equity holder in publicly-traded company. Ukropec:Janssen: Current Employment, Current equity holder in publicly-traded company. Qi:Janssen: Current Employment, Current equity holder in publicly-traded company; Johnson and Johnson: Current equity holder in publicly-traded company. Lin:Janssen Scientific Affairs: Current Employment, Current equity holder in publicly-traded company. Richardson:Celgene/BMS, Oncopeptides, Takeda, Karyopharm: Research Funding.
The specific regimen combination is not yet approved, but individual components are.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal